The XV Collection: Anatomy of a Protein Kinase Spine and How to Break It

by Ann Stock
The post-translational addition of phosphate groups to serine, threonine and tyrosine residues is a fundamental strategy for regulating protein activities in eukaryotes. Eukaryotic protein kinases — the enzymes that catalyze these modifications — are critical to cellular function, and aberrant kinase activities are associated with many diseases including cancer, inflammation, infection, diabetes, hypertension, and neurodegeneration.
Eukaryotic protein kinases are therefore important targets for therapeutic intervention, now constituting a quarter of all drug discovery and development efforts, and ranking second only to G-protein-coupled receptors (GPCRs) as pharmaceutical targets. More than three dozen kinase inhibitors have received FDA approval since 2001, when Imatinib (Gleevec), an inhibitor of bcr-abl kinase, was approved for treatment of chronic myelogenous leukemia (CML). Rational drug design has played an essential role in kinase inhibitor development, with molecules being designed to target specific kinase conformations. Understanding the structural basis for regulation of protein kinase activity is therefore key to these efforts.
For the PLOS Biology XV Collection, I’ve chosen to highlight an article from the laboratory of Susan Taylor (Meharena et al., 2013) that defines a set of intramolecular interactions that distinguish inactive and active conformational states of eukaryotic protein kinases. Such classification is complicated, because unlike many enzymes, eukaryotic kinases do not have single discrete active and inactive conformations, but instead are dynamic, with multiple conformations populating the two functional states.
The catalytic core of eukaryotic protein kinases consists of conserved N- and C-lobes with the active site located at the interface of these two lobes. Previous studies had identified three hydrophobic features in the catalytic core: the αF-helix in the C-lobe and two clusters of non-contiguous residues in the primary sequence that coalesce in the three-dimensional structure to form two hydrophobic “spines” that span the N- and C-lobes. The Catalytic (C) spine includes the adenine ring of bound ATP, which bridges hydrophobic residues in the N- and C-lobes. The Regulatory (R) spine, which typically consists of two aromatic residues in the C-lobe (RS1 and RS2) and two aliphatic residues in the N-lobe (RS3 and RS4), runs parallel to the C-spine, is aligned in a contiguous hydrophobic patch in the active state, and is disassembled in the inactive state.
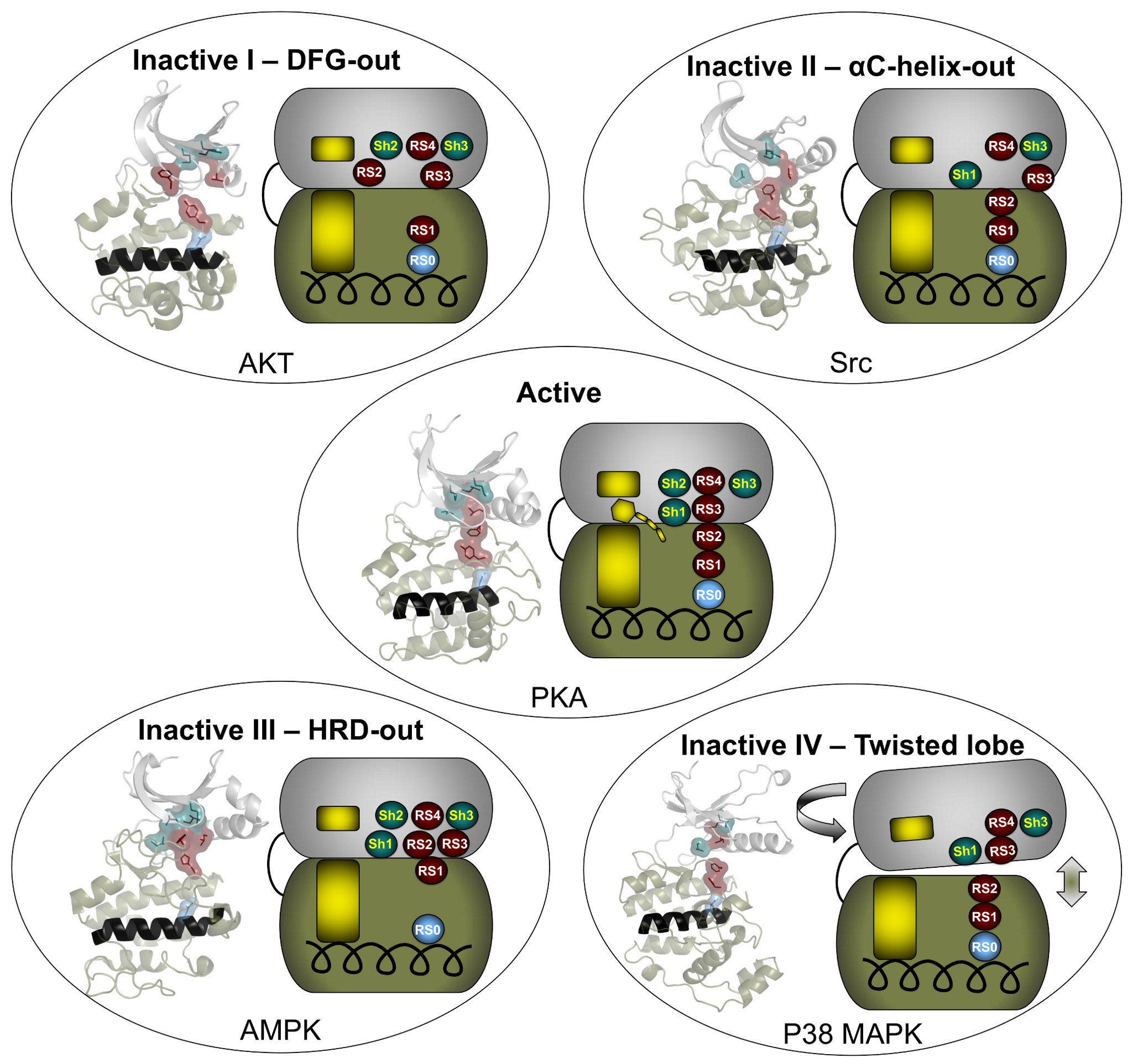
Meharena and colleagues examined R-spine residues in the sequences of ~13,000 kinases and tested hypotheses about the nature of these residues by mutational analyses of the representative kinase cAMP-dependent protein kinase (PKA). They observed that residues RS1 and RS2 in the C-lobe were extremely sensitive to mutation, in contrast to the relative robustness of residues RS3 and RS4 in the N-lobe. This led to the identification of a set of three highly conserved residues that surround RS3 and RS4. These residues that support the R-spine in the N-lobe were dubbed Shell residues Sh1, Sh2 and Sh3. Additional mutational analyses provided experimental validation of the hypothesis that integrity of the R-spine is essential for catalysis. Furthermore, the data provided evidence that phosphorylation of the activation loop promotes activation by stabilizing the R-spine.
Knowing that an assembled R-spine is required for an active state, the group examined available structures of eukaryotic protein kinases and identified 172 structures in which R-spines were disassembled. They were able to classify four specific ways in which the R-spine was broken. Two of these correlated with previously characterized inactive conformations associated with positioning of the DFG motif in the activation loop. One conformation involves a DFG-out orientation, and the other, a DFG-in orientation caused by movement of the αC-helix. These two inactive conformations have already been successfully targeted for drug development.
The description of additional inactive conformations provides opportunities for new strategies for drug design and a broader foundation for interpreting and perhaps eventually modulating the molecular defects caused by protein kinase mutations associated with human disease.
Meharena HS, Chang P, Keshwani MM, Oruganty K, Nene AK, Kannan N, Taylor SS, Kornev AP (2013) Deciphering the structural basis of eukaryotic protein kinase regulation. PLOS Biology 11(10): e1001680. https://doi.org/10.1371/journal.pbio.1001680
Ann Stock is at the Center for Advanced Biotechnology and Medicine and the Robert Wood Johnson Medical School Department of Biochemistry, Rutgers University, and is a member of the PLOS Biology Editorial Board.

This blog post is the seventh in a series of twelve, forming PLOS Biology’s XV Collection, celebrating 15 years of outstanding open science; read Lauren Richardson’s blog for more information.
Featured image credit: pbio.1001680
Ann Stock image credit: Ann Stock